
In this learning activity, we will review the GALA study's high-level findings to assist practitioners to understand the presentations, diagnosis and prognosis of Alagille Syndrome.
Alagille syndrome (ALGS) is a rare, life-threatening, multisystem disorder that primarily affects the liver, with significant cardiac, renal, neurological, vascular, and ophthalmic invovlement. The estimated prevalence is 1 in 30,000 live births, although various reports suggest that the incidence may be higher than previously reported. 1, 2, 3
ALGS follows an autosomal dominant inheritance pattern caused by pathogenic variants in JAGGED1 (JAG1) and NOTCH2, both of which are critical components of the Notch signalling pathway. JAG1 mutations account for approximately 94.3% of cases, while NOTCH2 mutations are implicated in 2.5% of patients. 8,9,10
Liver disease in ALGS typically presents early, usually within the first three months of life.
It is characterised by high γ-glutamyl transferase cholestasis, which can lead to pruritus, growth deficits, the development of xanthomas, and progressive liver failure. 4
Cholestatic pruritus is particularly debilitating, affecting up to 88% of children and often necessitating liver transplantation, even without hepatic failure. Additionally, xanthomas, occurring in 24–42% of patients, pose a significant disease burden and are a common factor in the decision for transplantation..5
Beyond the liver, ALGS presents a broad spectrum of extra-hepatic manifestations. Cardiac anomalies, present in 97% of cases, range from benign murmurs to complex congenital heart defects, which are strong predictors of early mortality.6
Ophthalmic abnormalities, particularly posterior embryotoxon, and renal complications, such as renal dysplasia, are also common. Additionally, characteristic facial features and butterfly vertebrae are frequently observed, though they are not essential for diagnosis. 6,7
ALGS exhibits significant variability in disease progression, and no genetic predictors for liver disease severity have been identified.
The natural history of ALGS remains incompletely understood, with existing data often derived from single-centre studies lacking molecular characterization. Prior to this study, the most comprehensive study was conducted across North American tertiary liver centres. It primarily included severely affected patients, leaving gaps in understanding the full spectrum of ALGS-related liver disease. The lack of data from developing countries further complicates this situation.8,7
The Global Alagille Alliance (GALA) Study Group was established to examine liver disease progression in a diverse, global cohort of children with ALGS. Its goals include assessing native liver survival (NLS), identifying early laboratory predictors of long-term outcomes, and evaluating post-transplant survival rates in a large international cohort. These insights are crucial, as targeted treatments have been shown to improve patient outcomes significantly.
Notch signalling is an evolutionarily conserved pathway that plays a critical role in the development and homeostasis of multiple tissues and organs. Aberrations in this pathway can lead to both cancerous and noncancerous diseases.10
The NOTCH1 gene provides instructions for the Notch1 protein, a receptor that interacts with specific proteins called ligands. When a ligand binds to Notch1, it sends important signals that guide the normal development of various tissues in the body, both before and after birth. Notch1 signalling helps determine how cells specialize into different types, as well as their growth, division, and self-destruction.10
Similarly, the NOTCH 2 gene codes for the Notch2 receptor, which also binds ligands to send signals crucial for developing tissues like the heart, liver, kidneys, and bones during embryonic growth. After birth, Notch2 plays a role in immune function, tissue repair, and bone remodelling, ensuring the body maintains healthy bone structure11
Understanding the function of Jag1 and its interactions within the Notch signalling pathway is crucial for exploring potential therapeutic targets for conditions related to ALGS and other developmental disorders.
Study Purposes
This study reviews the high-level findings of the GALA study and is intended to assist practitioners in understanding the presentations, diagnosis and prognosis of Alagille.
Study Methodology
The GALA Study was established in 2018 and is ongoing. It currently includes 67 paediatric centres across 29 countries. The initial findings of the study were recorded in this study under review here.
Children with clinically or genetically confirmed Alagille syndrome who were born between January 1997 and August 2019 were included. Furthermore, children born before 1997 who had a pathogenic or likely pathogenic variant in JAG1 or Notch2 were eligible for inclusion.
The pathogenicity of reported variants was classified according to the American College of Medical Genetics and Genomics guidelines.12
Key study parameters included neonatal cholestasis and liver involvement, with variables such as biochemical parameters and cardiac involvement assessed through various diagnostic reports.
The cohort was stratified geographically as: Africa, Asia, Europe, the Middle East, North America, Oceania (Australia and New Zealand), and South America.
Causes of death were classified into one of the following categories: liver or liver-related complications, cardiac-related complications, noncardiac vascular complications, multiorgan failure, sepsis, bleeding, other, or unknown.
Key Study Findings
The GALA study included 1,433 children with clinically or genetically confirmed ALGS who were followed up for a median of six years. 57% of the patients were male.
The largest patient cohort was from Europe (34%), followed by North America (29%) and Asia (23%).
Molecular genetic testing was available in 62% of patients with ALGS, and a pathogenic variant in JAG1 or NOTCH2 was identified in 98% of the patients.
Among genotyped patients, 2% were negative for a pathogenic variant in JAG1 or NOTCH2 despite meeting clinical criteria for ALGS (≥3 clinical characteristics).
The majority of ALGS cases were de novo (56%).
A competing risk analysis was conducted to evaluate native liver survival, liver transplantation, and the risk of mortality without LT. We have summarised the findings accordingly.
Native Liver Survival and Transplantation Outcomes in ALGS
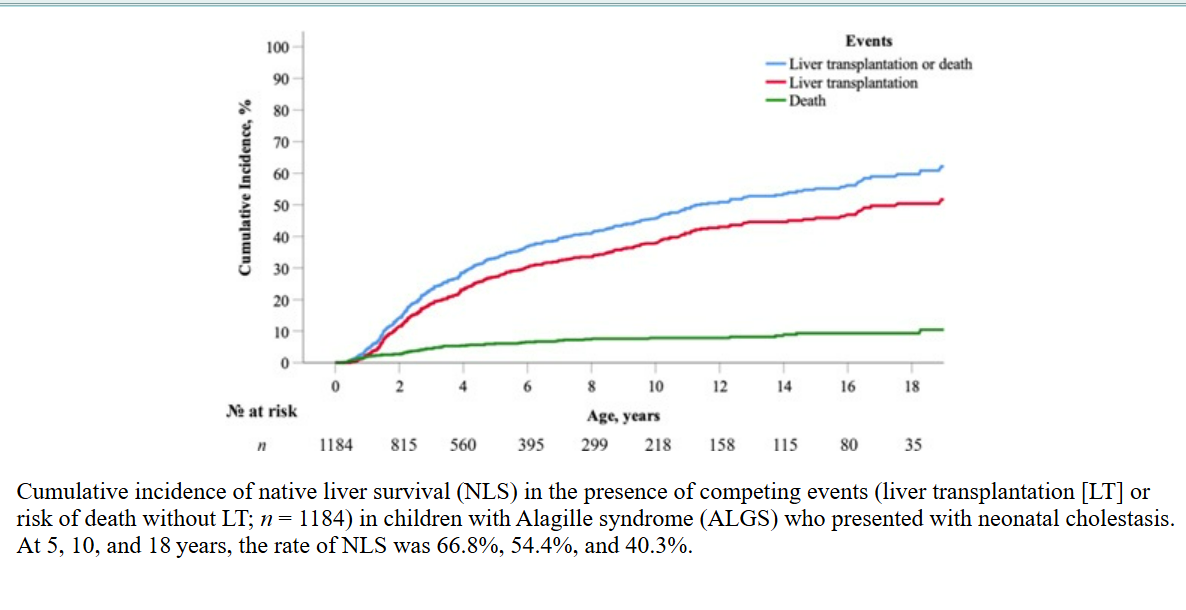
Among 1,184 children with a history of neonatal cholestasis, 345 underwent isolated liver transplantation (LT), while an additional four patients received combined liver-kidney transplantation.
A further 14 patients without documented neonatal cholestasis also underwent LT.
NLS rates declined over time, with 66.8% at 5 years, 54.4% at 10 years, and 40.3% at 18 years. The cumulative LT incidence increased to 27.1%, 37.8%, and 50.4% at the same intervals.
The risk of death without LT was 6.1% at 5 years, 7.8% at 10 years, and 9.3% at 18 years. Notably, no significant differences in NLS rates were observed between male and female patients.
No significant differences in NLS were observed between sexes, but rates varied across geographic regions.
Liver-Related Events and Progression of Portal Hypertension in ALGS
By the age of five, 36.3% of children had experienced at least one event, the incidence rising to 51.5% by 10 years and 66.0% by 18 years of age
Clinically evident portal hypertension (CEPH) was observed in 39.2% of children by 10 years and 65.8% by 18 years, based on ultrasound-confirmed splenomegaly and platelet counts below 150 × 109/L.
By adulthood, 22.3% required diuretics for ascites, and 12.7% developed varices necessitating endoscopic intervention.
Overall, 68.9% of patients developed CEPH, highlighting the progressive nature of liver disease in ALGS and the need for ongoing surveillance and early intervention.
Serial laboratory measurements were obtained for 605 patients across 3,777 follow-up visits during their first year of life.
Key laboratory markers—including total bilirubin (TB), direct bilirubin (CB), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and the AST to platelet ratio index (APRI)—demonstrated significant log-linear relationships with liver transplantation (LT) and mortality.
The study found the following:
Bilirubin thresholds were particularly predictive of native liver survival (NLS).
Among children aged 6 to 12 months, those with TB levels <5.0 mg/dL exhibited the highest NLS rate at 79.0%.
In contrast, survival rates significantly declined with TB levels ≥5.0 mg/dL (31.6%) and ≥10.0 mg/dL (18.2%) (p < 0.001).A TB level between 5.0 and 10.0 mg/dL was associated with a 4.8-fold increased risk of LT, while levels ≥10.0 mg/dL elevated this risk 15.6-fold.
Additionally, TB levels between 5.0 to <10.0 mg/dL indicated a 4.1-fold higher risk of clinically evident portal hypertension (CEPH).
AST levels ≥152 U/L also predicted LT risk (HR, 1.9; 95% CI, 1.4–2.6).
Sensitivity analysis confirmed that missing TB data did not significantly impact NLS rates (p = 0.78).
Hepatocellular Carcinoma in the Study Cohort
Histologically confirmed hepatocellular carcinoma (HCC) was identified in less than 1.0% of the study cohort (n = 9 out of 1,433), with a median age at diagnosis of 4.1 years (IQR, 1.5–8.3).
Hepatic fibrosis was present in 55% of the native liver biopsies (n = 5/9), and 75% of patients (n = 3/4) exhibited clinically evident portal hypertension (CEPH).
Among these individuals, six underwent liver transplantation (LT), two died prior to LT, and one patient had surgical resection and remained alive at the last clinical follow-up. Notably, the majority of HCC diagnoses occurred before LT (n = 4/6).
Mortality in Children with Alagille Syndrome (ALGS)
Throughout the study period, a total of 108 deaths were recorded among the 1,433 children diagnosed with Alagille syndrome (ALGS).
The survival rates at key intervals were notably high, with overall patient survival recorded at 92.8% at 5 years, 91.2% at 10 years, and 88.1% at 18 years.
However, significant variations in survival rates were observed across different geographic regions, indicating potential disparities in healthcare access and management (p = 0.002).
The median age at death for these patients was 2.6 years (IQR, 1.2–4.7), highlighting the early mortality associated with ALGS.
Among the causes of mortality, liver-related complications, including those stemming from liver transplantation, were the leading contributors, accounting for 22% of deaths.
The median age for deaths due to liver-related issues was 2.8 years (IQR, 1.6–6.7), underscoring the critical role of liver health in this patient population.
Following liver complications, cardiac-related issues were responsible for 18% of deaths, with a median age at death of 1.1 years (IQR, 0.5–4.5).
Additionally, both multiorgan failure and noncardiac vascular complications contributed to 15% of the mortality, with median ages of 3.9 years (IQR, 3.0–7.0) and 2.2 years (IQR, 1.5–3.2), respectively.
A comprehensive survival analysis was conducted to compare overall survival rates between children with ALGS who presented with a history of neonatal cholestasis (n = 1,184) and those without (n = 203).
The results revealed that children with a history of neonatal cholestasis had significantly lower cumulative survival rates at both 10 and 18 years compared to those without cholestasis (log-rank, p < 0.001).
Specifically, the survival rates at 10 and 18 years for patients with ALGS presenting with cholestasis were found to be 89% and 86%, respectively, whereas the rates for those without cholestasis were markedly higher, at 100% and 97%.
This discrepancy emphasizes the critical impact of neonatal cholestasis on long-term outcomes in children with ALGS.
Discussion
A key finding from the study is that 40% of children with ALGS who presented with cholestasis reached adulthood with their native liver. Notably, native liver survival (NLS) was influenced more by liver transplantation (LT) than by mortality, emphasizing the critical role of transplantation in disease management. However, significant geographic variations in NLS rates were observed, likely reflecting disparities in organ allocation policies and access to clinical resources. The median age for LT was 2.8 years, with most transplants occurring within the first five years of life due to severe cholestasis-related complications.
The median age for Liver Transplant in these patients was 2.8 years, with most transplants occurring within the first five years of life due to severe cholestasis-related complications.
Among the key prognostic indicators identified, serum total bilirubin (TB) levels between 6 and 12 months of age emerged as a reliable predictor of liver outcomes. Children with TB levels below 5.0 mg/dL demonstrated better NLS rates, providing a potential early marker for disease progression. In contrast, bile acid levels in the first year of life were not predictive of NLS, suggesting a gap in current clinical practice that may need to be addressed.
Histopathological findings further underscored diagnostic challenges in ALGS. Bile duct paucity, traditionally considered a hallmark of ALGS, was observed in only 65% of cases. This finding complicates differentiation from biliary atresia (BA) and highlights the need for improved diagnostic criteria.
Despite the complexity of ALGS, the study reports promising global survival rates, with overall survival exceeding 83% at 18 years. This figure reflects improvements in recipient selection, transplantation outcomes, and multisystem disease management.
However, early mortality post-LT remains a significant concern. The all-cause mortality rate of 8.5% in this cohort is lower than previously reported estimates, likely attributable to advances in LT availability and multidisciplinary care. Nonetheless, with most deaths occurring in early childhood, there is a growing need to extend awareness of ALGS into adult care settings. Currently, specific management guidelines for adult ALGS patients are lacking, underscoring the importance of long-term follow-up strategies.
This study reinforces the need for further research into ALGS, particularly in refining prognostic markers, optimizing transplantation criteria, and addressing gaps in clinical care for older patients.

- Native Liver Survival (NLS) and Transplantation Trends
- 40% of children with ALGS who presented with cholestasis reached adulthood with their native liver.
- Liver transplantation (LT), rather than mortality, was the primary factor influencing NLS.
- The median age for LT was 2.8 years, with most transplants occurring within the first five years due to severe cholestasis-related complications
- Geographic Variability in Outcomes
- Significant geographic differences in NLS rates were observed, reflecting disparities in clinical resources and organ allocation policies.
- Compared to the 24% NLS rate from the Childhood Liver Disease Research Network, the GALA cohort showed a higher 18-year NLS rate of 40%, suggesting differences in study populations and management approaches.
- Predictive Markers for Liver Disease Progression
- Serum total bilirubin (TB) levels in children aged 6–12 months were identified as a key prognostic marker, with levels <5.0 mg/dL associated with better NLS rates.
- Bile acid levels in the first year were not predictive of NLS, highlighting a gap in current clinical practice and potential limitations in monitoring disease progression.
- Diagnostic Challenges and Histopathology
- Bile duct paucity, a traditional hallmark of ALGS, was present in only 65% of cases, complicating differentiation from biliary atresia (BA) and suggesting a need for improved diagnostic criteria.
- Survival Rates and Need for Long-Term Care
- Overall survival exceeded 83% at 18 years, reflecting improved transplant outcomes and multisystem disease management.
- However, early post-LT mortality remains a concern, and most deaths occur in early childhood.
- There is an urgent need for adult ALGS care guidelines, as long-term disease management remains underexplored.
In Conclusion
The GALA study offers the most extensive real-world analysis of Alagille syndrome (ALGS) liver disease, examining over 1,400 children from 29 countries.
Findings suggest that total bilirubin (TB) <5.0 mg/dL between 6 and 12 months of age is associated with better native liver survival (NLS) and may help guide timing of liver transplantation (LT)—a particularly complex decision in ALGS.
With only 40.3% of children with ALGS reaching adulthood with their native liver, these natural history data are critical for evaluating emerging therapies currently in clinical trials. The study underscores the need for adult hepatologists to recognize ALGS and its multisystem complexities as more patients transition into adulthood.
These findings provide clinicians with an objective tool for treatment decisions and assessing new therapeutic interventions in ALGS.
To help us unpack the challenges of diagnosing and managing ALGS, we’re joined by Dr Marisca Baretta, a Paediatric Hepatologist at Wits University’s Donald Gordon Medical Centre.
Dr Baretta will walk us through why ALGS is frequently overlooked, how it impacts young patients, and what can be done to improve early detection and care. Stay with us as we explore the realities of rare liver diseases and what they mean for patients, families, and healthcare providers.
Access the Podcast Here
References
Publication Information
Published: 27 February 2025
Catalogue Number: MAICPD002
Category: Rare Diseases
Sub-Category: Rare Liver Diseases
Fact-Checked: 26 February 2025
Disclaimer
Every effort has been made to attribute quotes and content correctly. Where possible, all information has been independently verified. The Medical Education Network bears no responsibility for any inaccuracies which may occur from the use of third-party sources. If you have any queries regarding this article contact us
Fact-checking Policy
The Medical Education Network makes every effort to review and fact-check the articles used as source material in our summaries and original material. We have strict guidelines in relation to the publications we use as our source data, favouring peer-reviewed research wherever possible. Every effort is made to ensure that the information contained here accurately reflects the original material. Should you find inaccuracies or out-of-date content or have any additional issues with our articles, please make use of the Contact Us form to notify us.
